

























































































 免費參與·100+跨境活動
免費參與·100+跨境活動 
 免費下載·4000+跨境資料
免費下載·4000+跨境資料 
 免費學習·2000+直播課程
免費學習·2000+直播課程 
 免費加入·15萬+賣家交流群
免費加入·15萬+賣家交流群 


2020-04-03 10:26

首先我們來關注一下最近鬧得沸沸揚揚關于荷蘭購買口罩質量問題事件。2020年4月2日,外交部發言人華春瑩主持例行記者會,回應荷蘭采購口罩“質量問題”。此外,商務部也披露了核實結果:為個人防護口罩,企業出口前已做說明。
問:近日,荷蘭、比利時等部分歐洲國家媒體報道稱,從中國購買的口罩不合格,存在質量問題。中方對此有何評論?
答:關于荷蘭媒體稱口罩存在質量問題,根據中方有關部門初步調查了解,那批口罩是荷蘭代理商自己采購的, 中方企業發貨前已告知荷方此批口罩為非醫用口罩,出口報關手續也是以“非醫用口罩”名義履行的。
在當前全球抗疫形勢下, 中方急各國之所急,克服自身困難,有些企業加班加點,夜以繼日,積極為國際社會提供各種防疫物資。我們一貫高度重視出口產品質量。
中方有關部門剛剛出臺更加嚴格的監管措施,要求有關醫療物資出口企業在向海關報關時,必須提供書面或電子聲明,承諾出口產品已取得我國醫療器械產品注冊證書,符合進口國(地區)的質量標準要求。(出口新規詳情查看:美國突然取消對中國KN95口罩標準認可!4月1日起醫療物資出口有新規!附CE/FDA認證資格認證指南)
口罩分為不同的防護標準和等級,也有日用防護和專業醫用之分。當前,各國急需防疫物資,我們善意提醒使用方在購買和使用之前仔細核對產品用途和使用說明,以及是否符合采購方的使用標準,避免急中出錯,誤將非醫用口罩配用于醫用。
個別媒體在未弄清事實之前, 炒作所謂的中國產品質量問題,是不負責任、我希望他們不是別有用心的,因為這樣做不利于國際抗疫合作。
提醒:海關已通知企業按照《商務部 海關總署 國家藥品監督管理局公告2020年第5號 關于有序開展醫療物資出口的公告》要求,對出口醫用口罩、醫用防護服做出明確的質量監管措施,為落實好監管要求,海關要求各報關企業嚴格按照申報要求,在商品規格型號欄詳細填報品牌、規格型號和用途,達到醫用標準的必須填報醫用。如發現商品實際屬性為醫用,填報為非醫用情況,海關將按照不如實申報處理,請報關企業提醒“收發貨人”落實好主體責任。
4月2日,商務部召開例行新聞發布會,商務部外貿司二級巡視員(副司級)劉長于就荷蘭媒體報道從中國進口的口罩出現質量問題做出回應。
劉長于說,荷蘭公司向中國相關企業采購的這批口罩為個人防護用的非醫療用口罩。有關企業出口時也做了說明:非醫用口罩不能用于醫療用途,也不能用于在重癥監護室工作的醫護人員。
3月28日荷蘭一些媒體報道稱,荷蘭從中國購買的60余萬只口罩存在質量問題,被衛生部全部召回。
劉長于透露說,商務部注意到荷蘭媒體相關報道,對此高度重視,第一時間向地方商務主管部門、相關出口企業進行多方核實。
劉長于說,“根據有關材料,我們了解到,荷蘭公司向我相關企業采購的這批口罩為個人防護用的非醫療用口罩。有關企業出口時也做了說明。非醫用口罩不能用于醫療用途,也不能用于在重癥監護室工作的醫護人員。”
劉長于表示,中國政府一貫高度重視醫療物資質量安全,對相關產品實行嚴格管理。在疫情防控的特殊時期,為進一步加強醫療物資出口質量監管,規范出口秩序,商務部會同海關總署、藥監局發布公告,相關醫療產品出口必須取得我國醫療器械產品注冊證書,符合進口國(地區)的質量標準要求。
劉長于也強調說:“我們希望國外采購方選擇在我國藥監部門注冊的產品供應商,并在產品使用前進行相應的質量檢驗,嚴格按照產品適用范圍和操作規程正確使用。如在采購和使用中出現有關問題,建議雙方企業按商業化原則妥善協商解決。”
注意
近日,歐洲安全聯盟(European Safety Federation)在其官方網站上發布文章表示,他們從不同渠道獲悉“證書”用作PPE(包括FFP2/FFP3口罩)CE標記的依據,而這些“證書”沒有法律價值,不能用作合格評定的結論。到目前為止,已經在CELAB、ICR Polska、ISET、ECM、NPS、CIC、Amtre Veritas和GTS的信頭上看到了“證書”,目前尚不清楚這些文件是否真的是由上述組織簽發的。
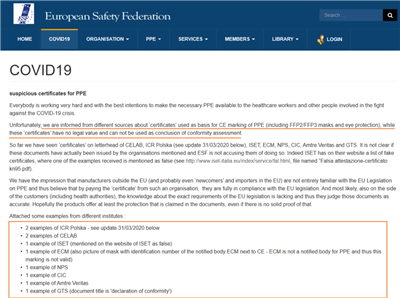
以下是來自不同機構的一些案例:
ICR Polska,2例
CELAB,2例
ISET,1例(已在ISET的網站上提到為假)
ECM,1例(口罩的CE標識旁邊有公告機構ECM的識別號,但ECM不是個人防護用品的公告機構,因此該CE標識無效)
NPC,1例
CIC,1例
Amtre Veritas,1例
GTS,1例(文件標題是自我聲明)
上述案例詳情及具體圖片,請查看歐洲安全聯盟官網下的案例鏈接。
https://www.eu-esf.org/covid-19/4513-covid-19-suspicious-certificates-for-ppe
歐洲安全聯盟表示,雖然目前當務之急是盡可能多地將口罩(和其他相關的個人防護用品)帶入歐盟,以便保護醫療工作者。但不可接受的是不提供聲稱的、保護低于標準的口罩提供給了現在處于高風險的醫護人員。根據(EU)2016/425號法規,防護面罩(如FFP2/FFP3)為III類PPE。這意味著合格評定包括:
1、由公告機構(Notified Body)進行的型式檢驗,合格后獲發“EU型式檢驗證書”,簡稱Module B證書。
2、由公告機構進行的生產跟蹤或隨機抽查或體系審核,簡稱Module D證書或Module C2證書。
所以,目前只有獲得歐盟 (EU) 2016/425 PPE法規口罩產品Module B、Module C2和/或Module D授權的認證公告機構才有權從事PPE個人防護口罩的CE認證。
鑒于疫情期間的PPE資源短缺及健康危機,歐盟委員會公布了關于合格評定和市場監督的(EU)2020/403號建議,允許成員國可以將個人防護用品投放到合格評定程序尚未完全完成的市場上。這僅適用于在危機期間由衛生保健主管部門購買的,但這并不意味著產品不必符合PPE法規中規定的適用基本健康和安全要求,進入正常銷售鏈的個人防護用品仍然必須完全符合規定。
歐洲安全聯盟提醒各相關利益方:檢查您收到的個人防護用品“證書”是否正確命名為“EU type examination certificate (EU型式檢驗證書)”,以及它們是否由合格的公告機構頒發,公告機構的識別號必須包含在證書中。
在任何情況下,請查看“EU type examination certificate (EU型式檢驗證書)” 的措辭(或另一種歐盟語言中完全相同的措辭——請查看國家版本的立法,以獲得正確的法律術語)。‘verification of compliance’, ‘certificate’, ‘certification report’等名稱都不是正確的法律術語,因此具有此類名稱的文件不是有效的EU型式檢驗證書。
如果上述“公告機構”的地址不在歐盟范圍內,這已經強烈表明文件存在問題,因為個人防護用品的公告機構都設在歐盟成員國或有相互承認協議的一些國家。
還要查找公告機構的名稱和編號(編號為4位)。為了確保公告機構確實是真實的且有相應資質的,您可以查看Nando數據庫進行辨別。
歐洲安全聯盟官網會定期更新PPE可疑證書的案例,詳情可查看:https://www.eu-esf.org/covid-19/4513-covid-19-suspicious-certificates-for-ppe
出口歐盟CE認證或將改為MDR
新冠疫情爆發后,口罩作為防治疫情的必需品,從在特定領域中使用的用品,而一躍變為了現實中一罩難求的日用必需品。
中國作為世界工廠,一夜之間也蜂擁出現了幾千家的口罩生產企業。這些口罩企業有相當多的瞄準了目前疫情嚴重的歐盟地區,希望將其的口罩投入歐盟市場,而歐盟市場的基本認證要求就是CE認證,它是打開并進入歐洲市場的“通行證”,是歐盟法律對限制類產品提出的強制性要求。
目前市場上新近完成的醫用口罩CE證書基本上都是基于歐盟醫療器械指令MDD 93/42/EEC進行發放的。
然而這其中存在一個潛在的危機:市場上部分口罩的CE證書,可能還有1個多月就要換版了。
關于MDR (EU 2017/745)分析
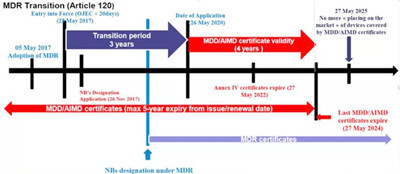
2017年5月5日歐盟就發布了新版醫療器械法規MDR(EU 2017/745)。在2017年5月25日,MDR正式生效。老的醫療器械指令即MDD( 93/42/EEC)與新的MDR(EU 2017/745)指令的交替過渡期為三年。
也就是說從2020年5月26日, MDR指令在歐盟就將開始強制執行,它將完全取代過去老的醫療器械指令MDD (93/42/EEC)和老的有源植入醫療器械指令AIMDD(90/385/EEC)。
但是,對于已經在歐盟渠道正式上市的產品來說,老的MDD指令CE證書可以保持到2024年5月26日;
需要特別指出的是
? MDR強制執行后,新申請的CE認證必須按照MDR執行;
? 當前沒有CE證書的產品,自5月27日起,必須按照MDR認證;
? 2020年5月26前簽發的MDD證書,在有效期內仍然可以用,最晚到2024年5月26日;
? 原有MDD證書需在證書失效前換發 MDR。
具體歐盟CE的醫療指令過渡時間安排如下圖所示:
5月26日起施行的歐盟MDR指令,對目前醫用口罩的CE認證,具體會有哪些影響?
1. 本次新冠疫情爆發期間所獲CE認證的醫用口罩,可以說95%以上的都是按照老的MDD指令進行的,可能要面臨新版換證問題;
2. 根據目前歐盟最新統計,擁有老指令版本MDD(93/42/EEC)授權的NB公告機構共有56家,而符合MDR授權的NB公告機構目前則僅有12家而已。也就是說,從2020年5月26日開始,針對醫用口罩的CE認證審核機構可選性降低了80%;
3. 由于歐盟MDR此類授權審核機構(NB:Notified Body)可選性的減少,必然造成醫用口罩CE認證費用相當大的概率將有大幅提升的可能;
4. 新版MDR指令審核要求比老版MDD指令更為復雜,認證周期必然大幅度拉長,本次疫情期間有些機構聲稱的幾天出證的可能性將基本為零;
5. 獲得CE認證的口罩等醫療設備必須要求有指定的歐盟授權代表(簡稱"歐代"),這在以前MDD指令時對一些低風險產品其實有沒有歐代監管并不嚴格,但在MDR指令后,即使是在一些電商平臺上進行銷售的醫用產品也會要求提供必要的歐代信息;
6. MDR的體系審核流程和要求更為復雜與繁瑣,舉個例子如在MDR條款15中要求,醫療器械廠商應在其組織架構內,至少配備一名負責監管合規的人員,即合規負責人(Person responsible for regulatory compliance)。該人員應具備醫療器械領域的必要專業知識,并且有一系列的資格性證明要求(如法律、醫學、藥學、工程或其他相關學科,并且至少擁有一年與醫療器械法規事務或質量管理體系相關的專業經驗);
7. 對于已經在歐盟渠道正式上市的產品來說,老的MDD指令CE證書可以保持到2024年5月26日。但如果企業產品在今年5月26日前,并未在歐盟市場銷售的,原則上在今年5月26日后應該將老的MDD證書重新申請調整到MDR版本。
此次歐盟是直接發布的Regulation(法規),相比較之前的Directive(指令)其區別在于:提高了約束力,發布立即在歐盟成員國生效并成為有約束力的法律,此次的Regulation無需向Directive那樣需要經過成員國轉化成當地法律法規去落實實施。
因此,企業在申請醫療產品CE認證時,在過渡階段請謹慎考慮是選用最新法規還是采用老的指令方案,同時也需要對NB機構的發證資格進行了解和確認以保證產品在歐盟市場銷售的可延續性。
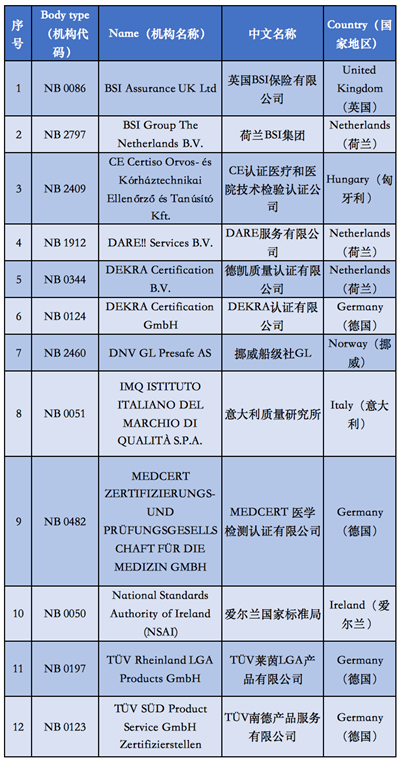
附①:2020年5月26日起
12家MDR授權機構清單
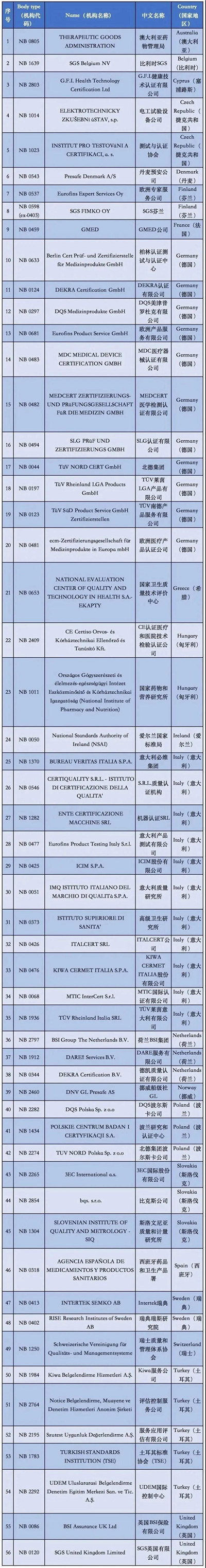
附②:2020年5月26日前
56家MDD授權機構清單
受疫情影響,很多國家和機構的政策都是頻繁變動的,比如美國對于中國標準KN95口罩的態度,所以,MDR指令是否會實施,是否會延期實施,目前都還是未知數,只能靜觀其變!
歐盟CE認證資格指南
出口歐盟市場,CE認證必不可少。目前市場上出現了各色各樣的醫療CE證書,讓人眼花繚亂。在各種行業微信群里,經常可以看到有人發出一張所謂的CE證書,請大家幫忙辨別真偽。為了更好地幫助到大家,下面我們就來談談具體的鑒別方法。
查詢CE證書的真偽,有多種方式,首先是最簡單粗暴的一種。大的公告機構會在自己的官網上開放查詢證書的窗口,當然,這種方式僅適用于發證機構正好提供了查詢服務的情況。而對于未開放證書查詢服務的機構,就不會奏效了。那么對于此類情況,當我們拿到一張醫療CE證書時,我們又該如何鑒別呢?
我們依然可以嘗試從您手上這張證書的發放機構入手,去歐盟官網查詢,看它是否具備歐盟醫療器械指令MDD 93/42/EEC或MDR醫療器械法規(EU) 2017/745及個人防護裝備授權(EU)2016/425的相應認證資質。
歐盟官網MDD 93/42/EEC醫療器械指令授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=13
歐盟官網MDR (EU) 2017/745醫療器械法規授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=34
歐盟官網 (EU)2016/425個人防護裝備授權的機構查詢地址:
https://ec.europa.eu/growth/tools-databases/nando/index.cfm?fuseaction=directive.notifiedbody&dir_id=15550
通過歐盟官網可以看到,擁有MDD 93/42/EEC醫療器械指令授權的公告機構共有56家,具體的機構清單,公告號,以及其有資質審核的產品范圍都詳細羅列在上面。
自2020年5月26日起,MDR (EU) 2017/745醫療器械法規將正式取代歐盟現行的MDD醫療器械指令強制實施,同樣在歐盟官網可以查詢到,擁有MDR授權的公告機構目前只有12家。
所以,如果您手上的醫療CE證書發證機構不在以上名單范圍內,則說明它并不具備醫療產品的歐盟發證資質,更別談CE證書的發放了,那么,很遺憾地說,您拿到的這張“”CE證書“是無效的。
另外,我們也可以從醫療器械產品CE認證的流程著手去分析,完成鑒別。
以口罩為例,首先,確認此口罩是否屬于醫療器械。口罩分為醫用口罩和防護口罩兩種,如果是后者,則不屬于醫療器械,不需要滿足歐盟醫療法規要求,按照PPE個人防護指令完成CE認證即可。如果是醫用口罩,則需要按醫療器械法規完成認證。而對于醫用口罩來說,則需要進一步確認它是否無菌。如果是無菌醫用口罩,在歐盟屬于一類帶滅菌醫療產品,必須按照醫療器械指令/法規MDD/MDR進行CE認證,這類情況是一定需要有授權的公告機構參與的。如果是非無菌醫用口罩,則是按照醫療器械指令/法規MDD/MDR進行CE自我宣稱,企業不需要通過公告機構認證,在準備好相應文件及測試報告等資料后,即可自行完成符合性聲明。
就目前的情況而言,鑒于無菌醫用口罩CE認證的難度較高且需要的時間較長,絕大部分的廠家都選擇了非無菌醫用口罩來生產和完成認證。
這里需要劃個重點,既然是由制造商進行自我宣稱CE符合性,又何來公告機構發放CE證書一說呢?如果不能發放CE證書,那很多企業所拿到的所謂證書又到底是什么呢?讓我們找一些模板來看看:

請大家仔細研究一下其內容:“Verification of the presence of the Technical Files in regards of the Medical Devices Directive…” 意思是此證書證明了機構復核過該企業已經按照醫療器械法規要求來準備技術文檔。
再來:“This document has been issued on voluntary basis and not as NB…” 意思是此證書是自愿性出具,不代表我以公告機構名義來執行了此事。
“…declares that the only scope of the assessment is to verify the existence of the declaration issued by the manufacturer or an applicant under its own responsibilities” 意思是此證書只是從他們的角度核實了制造商或申請人根據其本身的責任所發出的符合性聲明是存在的。
因此,這類所謂的證書不是真正意義上由有授權的公告機構出具的CE證書。所以請大家務必擦亮眼睛鑒別真偽。
美國FDA認證應急授權
美國對于醫療器械的管理歸口在美國食品藥品監督管理局(FDA)的器械和輻照健康中心(CDRH)。醫用口罩、體溫計(包括耳溫槍、額溫槍、普通電子體溫計和水銀體溫計)和在中等至高等風險下使用的防護服和隔離服都是二類醫療器械,需要申請510(k)。N95口罩雖然可以豁免510(k),但必須首先取得美國疾控中心NIOSH的N95證書。美國FDA對于所有的醫療器械僅有處方和非處方之分,并沒有家用和專業醫護人員使用的區別。
510(k)的申請流程如下:
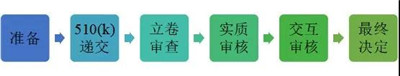
通常一個產品從啟動510(k),準備測試和各類文件,直至最終的審核結束,需要歷經8-10個月,而初次申請的企業,這個時間通常會更長。
除了510(k)以外,FDA要求所有的醫療器械企業都需要進行場所注冊(Establishment Registration)和產品列示(Product Listing),這一要求對于應急使用授權的產品也不例外。對于所有的海外企業,在進行場所注冊之前應先獲得鄧白氏編碼,編碼在中國由華夏鄧白氏公司代理發放,免費的代碼需要大約30天可以獲得。獲取鄧白氏編碼后,大約需要1-2周左右完成場所注冊和產品列示。
無論是510(k),場所注冊或者產品列示,FDA都不會向企業頒發任何證書,僅以美國FDA數據庫中的數據為準。也就是說,大家見過的各種有著老鷹標記的證書都是沒有任何效力的。
目前,新冠疫情在美國呈現明顯爆發的趨勢,各類醫療物資也趨于緊張。美國食品藥品監督管理局(FDA)早在今年(2020年)2月初就開始對醫療器械潛在短缺的情況進行調查,為了應對各類醫療器械的緊缺FDA發布了各類應急使用授權(Emergency Use Authorization,EUA)。目前可以申請EUA的產品主要是未獲上市的N95口罩、未獲上市的新冠病毒診斷試劑、未獲上市的酒精洗手液產品、已上市但需擴展用途的非侵入遠程監護系統、已上市和未上市的呼吸設備。
新冠檢測試劑:首當其沖的就是診斷試劑,FDA已經就該產品發布了第二版的指導原則。第一版指導原則主要針對美國國內臨床實驗室自我開發的檢驗方法的EUA申請,而第二版則納入了針對生廠商的遞交EUA申請的詳細指導。EUA的申請要包含的內容與510(k)相似,需要提交檢測試劑的描述、預期用途、性能評價報告、臨床評價方案、穩定性試驗方案、標簽標識等。由于屬于應急審批,FDA還要求企業必須提供針對患者和專業人員的明白紙(Fact Sheet)。目前海河咨詢已經完成了新冠病毒膠檢測試劑盒膠體金法的EUA申請。
口罩:其次緊缺的就是口罩。口罩在美國有多種類別,海河也專門針對口罩注冊途徑舉辦過專題線上研討會,針對不同類別的上市途徑進行過說明。FDA也針對口罩發布了2輪的EUA通知。
第一輪的EUA允許直接使用滿足如下條件的口罩:
(1)根據NIOSH42 CFR part 84作為非動力空氣凈化過濾面罩口罩批準的所有一次性過濾式口罩呼吸器(包括N95口罩)。以及
(2)經NIOSH批準但已過制造商推薦的保存期限的過濾面罩口罩,供醫護人員在醫療環境中使用,以防止醫護人員由于面罩口罩短缺,而暴露于病原性生物空氣傳播顆粒中。
考慮到這樣的措施還是無法保障美國市場的口罩供應,FDA又在近期發布口罩的EUA申請,特別地,沒有獲得NIOSH批準的口罩也可以進行EUA的申請,但是必須符合:
條件1-滿足特定國家/地區性能標準,包括
澳大利亞:P2,P3
巴西:PFF2,PFF3
歐盟:FFP2,FFP3
日本:DS/DL3,DS/DL2
韓國:Special 1st
墨西哥:N100, P100, R100, N99, P99, R99, N95, P95, R95
和/或
條件2-獲得特定國家上市許可,包括:
歐盟:CE認證;
澳大利亞:ARTG注冊
加拿大:注冊證;
日本:醫療器械注冊證。
非侵入遠程監護系統:例如可穿戴的設備,手持設備或者家用固定監護設備。產品類別包括體溫計、心電圖機、心電軟件、血氧儀、血壓計、呼吸監測設備等。這些監護設備應當具有潛在的網絡連接能力,包括藍牙,Wi-Fi或蜂窩網絡等,并且能將檢測數據直接傳輸給醫療機構。為了應對新冠疫情,對于已經獲得FDA許可上市的非侵入遠程監護系統,在應急使用期間,FDA將允許其在適應癥,宣稱功能,硬件或軟件等各方面的有限的變更,而不需要對變更遞交510(k)申請。
呼吸設備:適用于應急使用授權的呼吸類設備指向具有呼吸衰竭或呼吸不足情況的病人提供通氣和呼吸支持的設備,包括但不限于醫院用連續呼吸機,家用連續呼吸機,應急使用呼吸機等。FDA仍然建議醫療機構使用FDA許可的傳統/全功能的呼吸機,但是考慮到盡可能多的可獲得性,對于已經獲得FDA許可上市的呼吸設備,在應急使用期間,FDA將允許其在適應癥,宣稱功能,硬件或軟件等各方面的有限變更,而不需要對變更遞交510(k)申請。此外,對于呼吸設備,FDA還開通了EUA申請。對于有興趣進行申請的生產商,FDA將提前介入與生產商溝通相關申請文件的要求以及對相關的申請進行加急審核。
酒精洗手液產品:之前酒精洗手液產品在美國是按照非處方類藥品進行管理的,洗手液中的酒精則是按照API進行管理的。目前,FDA已經發布通知,不會對提供用于洗手液的酒精的生產商或者洗手液的生產商采取行動,即可以認為,FDA將允許用于洗手液的酒精和基于酒精的洗手液在滿足限定條件下直接在美國銷售。海河咨詢特別提醒:對于洗手液的生產商僅限定適用于藥劑師以及聯邦設施,不適用于其他商業生產商。
(來源:海運信息)
以上內容屬作者個人觀點,不代表雨果網立場!如有侵權,請聯系我們。



























