





























































































 免費參與·100+跨境活動
免費參與·100+跨境活動 
 免費下載·4000+跨境資料
免費下載·4000+跨境資料 
 免費學習·2000+直播課程
免費學習·2000+直播課程 
 免費加入·15萬+賣家交流群
免費加入·15萬+賣家交流群 


2020-03-19 10:15

近期,全球疫情備受關(guān)注,很多外貿(mào)企業(yè)咨詢口罩出口的詳細要求及各國口罩準入條件,圈妹在此收集整理了海關(guān)總署及廣東省市場監(jiān)管局下屬的廣東省WTO/TBT通報咨詢研究中心等單位發(fā)布的相關(guān)資料,在此分享給各位。
主要內(nèi)容分為三部分:
1、口罩出口通關(guān)提示
2、口罩出口前準備
3、各國口罩準入條件
出口通關(guān)提示
報關(guān)前提條件:
收發(fā)貨人注冊編碼(慈善機構(gòu)可為臨時編碼),需辦理無紙化通關(guān)法人卡
出口資質(zhì):
口罩出口對生產(chǎn)銷售單位、境內(nèi)發(fā)貨人,除滿足國內(nèi)生產(chǎn)、市場流通資質(zhì)需求外,中國海關(guān)無特殊資質(zhì)要求。
出口申報要求:
1.商品歸類:除特殊情況外,絕大部分口罩應歸入稅號63079000。
2.檢驗檢疫:口罩為非法檢產(chǎn)品,申報時檢驗檢疫項目無需填報。根據(jù)我國政府與相關(guān)國家簽訂的政府間檢驗協(xié)議,對出口伊朗等少數(shù)幾個國家的產(chǎn)品需按規(guī)定進行裝運前檢驗。
3.關(guān)稅征免:如出口物資為貿(mào)易性質(zhì),征免性質(zhì)申報一般征稅,征免方式申報照章征稅;如為捐贈性質(zhì),境內(nèi)發(fā)貨人為貿(mào)易代理商、慈善機構(gòu)等,征免性質(zhì)可不填,征免方式申報全免。
4.禁限管理:目前商務部未對口罩設置貿(mào)易管制要求,中國海關(guān)也無針對防護物資的監(jiān)管證件口岸驗核要求。
5.申報規(guī)范:按照規(guī)范申報要求填寫商品名稱、成分含量;如物資非中國生產(chǎn),原產(chǎn)國按照實際生產(chǎn)國填寫。
出口退稅:
口罩的出口退稅率為13%。
中美關(guān)稅排除加征:
美國企業(yè)可申請排除口罩進口加征關(guān)稅,但是目前只有少數(shù)企業(yè)獲準豁免。詳見美國貿(mào)易代表辦公室網(wǎng)站https://ustr.gov/。
快速通關(guān)保障:
物資出口申報如遇單窗等系統(tǒng)故障,可聯(lián)系現(xiàn)場海關(guān)采取應急方式處置,或者撥打海關(guān)12360熱線進行咨詢。
出口前準備
以下內(nèi)容是根據(jù)國內(nèi)外相關(guān)政府機構(gòu)、專業(yè)網(wǎng)站、新聞報道收集整理而成,僅供參考。具體內(nèi)容以相關(guān)管理部門、國外官方機構(gòu)要求為準。
明確口罩分類:
國外按照用途一般分為個人防護和醫(yī)用兩類口罩。
國內(nèi)出口貿(mào)易企業(yè)需具備的資質(zhì)和材料:
1.營業(yè)執(zhí)照(經(jīng)營范圍有相關(guān)經(jīng)營內(nèi)容)。
2.企業(yè)生產(chǎn)許可證(生產(chǎn)企業(yè))。
3.產(chǎn)品檢驗報告(生產(chǎn)企業(yè))。
4.醫(yī)療器械注冊證(非醫(yī)用不需要)。
5.產(chǎn)品說明書(跟著產(chǎn)品提供)、標簽(隨附產(chǎn)品提供)。
6.產(chǎn)品批次/號(外包裝)。
7.產(chǎn)品質(zhì)量安全書或合格證(跟著產(chǎn)品提供)。
8.產(chǎn)品樣品圖片及外包裝圖片。
9.貿(mào)易公司須取得海關(guān)收發(fā)貨人注冊備案。
國內(nèi)出口口罩生產(chǎn)企業(yè)資質(zhì)證明:
生產(chǎn)個人防護或者工業(yè)用非醫(yī)療器械管理的普通口罩,有進出口權(quán)的企業(yè),可自行直接出口。
生產(chǎn)屬于醫(yī)療器械管理的口罩用于出口,中國海關(guān)不需要企業(yè)提供相關(guān)資質(zhì)證明文件,但一般進口國會要求生產(chǎn)企業(yè)提供產(chǎn)品三證,以證明該進口的商品在中國已合法上市,具體如下:
1.營業(yè)執(zhí)照(經(jīng)營范圍包含有醫(yī)療器械相關(guān),非醫(yī)療級別的物品不需要)。
2.醫(yī)療器械產(chǎn)品備案證或者注冊證。
3. 廠家檢測報告。
生產(chǎn)企業(yè)有進出口權(quán),可以自行出口,如沒有進出口權(quán),可以通過外貿(mào)代理進行出口銷售。
內(nèi)貿(mào)企業(yè)做出口需要取得的基本資質(zhì):
1. 向市場監(jiān)管部門取得營業(yè)執(zhí)照,增加經(jīng)營范圍“貨物進出口、技術(shù)進出口、代理進出口”。
2. 向商務部門取得進出口權(quán),可直接在商務部業(yè)務系統(tǒng)統(tǒng)一平臺(http://iecms.mofcom.gov.cn/)申請,網(wǎng)上提交材料。
3. 向外匯管理局申請取得開設外匯賬戶許可。
4. 辦理進出口貨物收發(fā)貨人海關(guān)注冊登記。
各國口罩準入條件(產(chǎn)品準入條件)
美國
1、必要資料:提單,箱單,發(fā)票。
2、個人防護口罩:必須取得美國 NIOSH檢測認證,即National Institute for Occupational Safety and Health美國國家職業(yè)安全衛(wèi)生研究所認證。
3、醫(yī)用口罩:須取得美國FDA注冊許可。
4、監(jiān)管及出口須知:
據(jù)路透社報道,美國貿(mào)易代表處3月12日宣布,不對部分從中國進口的醫(yī)藥品加征關(guān)稅。
這些醫(yī)藥品包括口罩、聽診器、血壓計袖帶等。
這一決定出于目前新冠疫情正在對美國健康醫(yī)療體系造成沖擊。此前美國貿(mào)易代表處已將洗手液、醫(yī)用手套等進口產(chǎn)品移出征稅清單。
① FDA監(jiān)管措施
FDA根據(jù)風險等級將醫(yī)療器械產(chǎn)品分為3個監(jiān)管控制類別,涵蓋近6000個產(chǎn)品代碼(product code),根據(jù)不同的風險采取相應的監(jiān)管力度。
不同類別醫(yī)療器械的監(jiān)管措施:
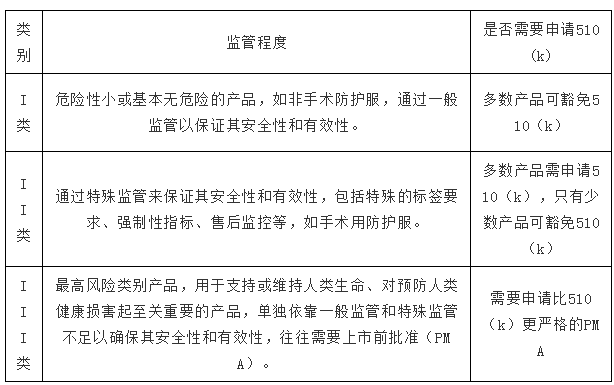
FDA 510(k)又稱上市前登記(Premarket Notification),是向FDA提交的上市前報告。FDA 510(k)的實質(zhì)是證明器械的實質(zhì)等同。
根據(jù)FDA的要求,少數(shù)I類和大部分II類醫(yī)療器械在美國上市前,至少需要提前90天遞交510(k)申請,用于證明要銷售的器械至少與已合法銷售的器械(即等價器械)一樣安全有效[21 CFR 807.92(a)(3)]。
在申請者收到FDA聲明器械“實質(zhì)性等同”(SE)的信件格式指令之前,不可在美國銷售該器械。
510(k)的申請材料必須包括諸如產(chǎn)品代碼、標簽、器械技術(shù)特性的總結(jié)、測試結(jié)果等其他的資料。510(k)沒有固定的表格或者模板,但是所有的信息必須符合《聯(lián)邦法規(guī)》第21篇第807節(jié)中的要求。
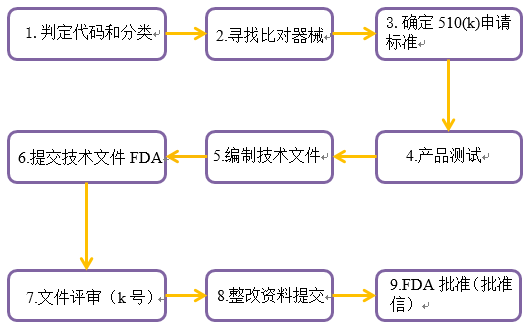
FDA 510(k)申請大致流程
如果FDA確定器械不是等價器械,申請者可以遞交另一份含有新數(shù)據(jù)的510(k)文件,提出重新分類請求,或者遞交上市前批準申批。
在FDA系統(tǒng)中對于口罩的分類代碼有如下3個。其中一個是外科口罩,一個是兒科口罩,一個是帶有抗菌/抗病毒介質(zhì)的外科口罩。
三個類別的口罩都屬于規(guī)則878.4040,分類都是Ⅱ類,都需要申請510(k)。
FDA不測試口罩,由申請者向FDA提供檢測數(shù)據(jù)和產(chǎn)品宣稱性能用于審核,檢測內(nèi)容包括顆粒過濾效率(PEF)、細菌過濾效率(BFE)、液體阻隔性、阻燃性等。510(k)申請周期較長,還有以下兩種可選路徑:
a)已經(jīng)獲得NIOSH批準的N95口罩可以直接注冊
可以看出,如果制造商的N95口罩獲得了NIOSH的批準,生物學測試、阻燃測試和血液穿透測試都通過了,那么則可以豁免510(k)的,可以直接進行工廠注冊和器械列名。
b)獲得持有510(k)的制造商的授權(quán),作為其代工廠使用其510(k)批準號進行企業(yè)注冊和器械列名。
申請方需要獲得授權(quán)書,需要簽署正式的質(zhì)量協(xié)議,F(xiàn)DA會進行核實和抽查。如果使用未經(jīng)許可的號碼,將會導致產(chǎn)品召回的風險。
② NIOSH認證
醫(yī)用口罩需要申請FDA 510(k)“上市前登記”。呼吸防護口罩則需要通過NIOSH認證,由NIOSH下屬的NPPTL實驗室實施認證。
口罩在出口美國之前,一般選擇先做NIOSH認證,再做FDA認證,產(chǎn)品將更受美國市場歡迎。如醫(yī)用N95口罩,需要既滿足NIOSH對于N95口罩的要求,同時也要滿足FDA相關(guān)標準。
NIOSH認證的流程如下:
1)提交申請書、技術(shù)資料(通常包括產(chǎn)品圖紙、產(chǎn)品說明、質(zhì)量體系文件、測試報告等)和樣品;
2)美國實驗室執(zhí)行測試與評估,NIOSH頒發(fā)證書和標簽;
3)生產(chǎn)商使用標簽。
NIOSH規(guī)定單獨的過濾式呼吸防護口罩上必須具有以下標識:
1)NIOSH認可的批準持有人/制造商名稱,注冊商標或申請人/批準持有人的企業(yè)名稱縮寫。如果適用,批準持有人對呼吸防護口罩進行私有標記的實體名稱可以代替NIOSH認可的批準持有人的業(yè)務名稱,注冊商標或批準持有人的業(yè)務名稱的縮寫;
2)NIOSH的大寫或NIOSH徽標;
3)NIOSH測試和認證批準號,例如TC-84A-XXXX;
4)NIOSH過濾特性和過濾效率等級,例如N95、N99、N100、R95、P95、P99、P100(目前NIOSH批準的過濾式呼吸防護口罩的七種類型);
5)型號或零件號:批準持有人的呼吸器型號或零件號,由一系列數(shù)字或字母數(shù)字標記表示,例如8577或8577A。
NIOSH建議還包括批號和/或生產(chǎn)日期,但這不是必需的。
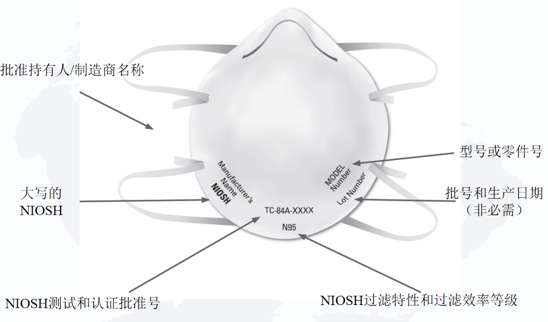
NIOSH過濾式呼吸防護口罩標識
歐盟
1、必要資料:提單,箱單,發(fā)票。
2、個人防護口罩:個人防護口罩的歐盟標準是EN149,按照標準將口罩分為FFP1/FFP2和FFP3三個類別。所有出口歐盟的口罩必須獲得CE認證證書。CE認證是歐盟實行的強制性產(chǎn)品安全認證制度,目的是為了保障歐盟國家人民的生命財產(chǎn)安全。
3、醫(yī)用口罩:醫(yī)用口罩對應的歐盟標準是EN14683。產(chǎn)品在歐盟銷售需要出具歐盟自由銷售證書 Free Sale Certificate,有了CE標志并進行了相關(guān)指令中要求的歐盟注冊后,中國的制造商出口歐盟不需要自由銷售證書。
4、監(jiān)管及出口須知:
① 呼吸防護口罩
呼吸防護口罩需要滿足法規(guī)(EU) 2016/425的要求,防護口罩屬于其中復雜設計的產(chǎn)品。出口歐盟需要授權(quán)的公告機構(gòu)(NB)進行認證并頒發(fā)證書,認證流程為:
a)提供申請表、產(chǎn)品實物圖片及說明書;
b)準備產(chǎn)品型式試驗報告。(依據(jù)EN 149檢測);
c)技術(shù)文件評審(由發(fā)證機構(gòu)評審);
d)工廠質(zhì)量體系審查(由發(fā)證機構(gòu)評審工廠體系資料);
e)公告機構(gòu)頒發(fā)CE證書。
② 醫(yī)用口罩
醫(yī)用口罩產(chǎn)品可分為無菌或非無菌狀態(tài),其認證模式不一樣。
非無菌狀態(tài)認證:
a)編制技術(shù)文件(TCF);
b)提供測試報告(依據(jù)EN 14683要求檢測,或提供熔噴布性能測試報告和無紡布生物學測試報告);
c)編制產(chǎn)品符合性聲明(DoC);
d)指定歐盟授權(quán)代表并完成歐洲注冊。
無菌狀態(tài)認證:
a)滅菌驗證;
b)建立ISO 13485醫(yī)療器械質(zhì)量管理體系;
c)編制技術(shù)文件(TCF);
d)提供測試報告(依據(jù)EN 14683要求檢測,主要提供細菌過濾效率、呼吸阻力、防濺阻力及滅菌驗證報告等);
e)公告機構(gòu)(NB)審核;
f)獲ISO 13485證書和CE證書;
g)指定歐盟授權(quán)代表并完成歐洲注冊。
從目前整體情況來看,如果之前沒有獲得公告機構(gòu)的CE證書,現(xiàn)在臨時申請周期比較長。因此,企業(yè)可考慮出口非無菌醫(yī)用口罩。但是非無菌醫(yī)用口罩并不是對生產(chǎn)環(huán)境完全無限制,EN 14683對于產(chǎn)品的初始污染菌要求不大于30 cfu/g。
日本
1、必要資料:提單,箱單,發(fā)票,日本國外的制造商必須向PMDA注冊制造商信息。
2、口罩包裝要求:
包裝上印有ウィルスカット(中文翻譯:病毒攔截)99%的字樣
PFE:0.1um微粒子顆粒過濾效率
BFE:細菌過濾率
VFE:病毒過濾率
3、口罩品質(zhì)標準:
1) 醫(yī)用防護口罩:符合中國GB 19083-2010 強制性標準,過濾效率≥95%(使用非油性顆粒物測試)。
2)N95口罩:美國NIOSH認證,非油性顆粒物過濾效率≥95%。
3)KN95口罩:符合中國GB 2626 強制性標準,非油性顆粒物過濾效率≥95%。
4、監(jiān)管及出口須知:
由于非工業(yè)用口罩沒有關(guān)于性能的鑒定規(guī)范,會造成口罩的標識和廣告的內(nèi)容差異較大而給消費者帶來巨大誤解的情況。
為此,日本口罩行業(yè)協(xié)會于2006年1月制定并實施了關(guān)于口罩的“標識和廣告自愿性標準”,并呼吁所有協(xié)會會員的口罩制造商從保護消費者的角度出發(fā),履行其社會責任。
日本全國口罩行業(yè)協(xié)會的會員標志如下:

JHPIA對口罩的標識和廣告的規(guī)定,不得在口罩的容器、外包裝以及廣告上聲稱以下內(nèi)容:
①聲稱具有醫(yī)療用品方面的功效和效果,聲稱具有醫(yī)藥品、藥妝、化妝品、醫(yī)療器械的功效和效果;
②缺乏依據(jù)聲稱口罩濾料的收集效率數(shù)值的標識(但是,在有依據(jù)的情況下,可在標識出檢測方法或者檢測機關(guān)的前提下標識該數(shù)值,收集效率最高為99%)。
統(tǒng)一框內(nèi)標識格式如下:

韓國
1、必要資料:提單,箱單,發(fā)票,韓國進口商營業(yè)執(zhí)照。
2、個人防護口罩標準:KF (Korean filter) 系列分為KF80、KF94、KF99
3、執(zhí)行標準規(guī)范:MFDS Notice No. 2015-69
韓國醫(yī)療器械準入的法規(guī)門檻,基本分類為I、II、III、IV類,持證為韓國公司(License holder),韓國收貨人需要到韓國藥監(jiān)局Korea Pharmaceutical Traders Association. 提前備案進口資質(zhì)(沒有不行)網(wǎng)址:www.kpta.or.kr。
4、監(jiān)管及出口須知:
根據(jù)《藥品事務法》,醫(yī)藥輔品是指與疾病的治療和預防有關(guān)的產(chǎn)品,并由食品藥品安全局局長指定的,用于治療,減少,治療或預防人類或動物疾病的紡織品和橡膠制品、對人體無害或不直接影響人體的產(chǎn)品以及用于滅菌,殺蟲劑和類似目的的產(chǎn)品以預防傳染病的產(chǎn)品。
在韓國,這一分類下的物品有:口罩(手術(shù)用、衛(wèi)生保健用)、用于保護、處置患處的產(chǎn)品(如:眼罩、繃帶、紗布等)、衛(wèi)生巾、口腔衛(wèi)生用品、直接用于人體外部消毒劑(如洗手液)等。
醫(yī)藥輔品上市/進口前需先向食品醫(yī)藥品安全評估院或地方食品醫(yī)藥品安全廳進行申請許可。
根據(jù)《醫(yī)藥輔品的批準、通知、評價規(guī)定》,在初次申請衛(wèi)生口罩的認可時,應提供測試結(jié)果作為支持數(shù)據(jù)。指定準藥物范圍-執(zhí)法20211001食品藥品安全部公告2019-86-2019年9月30日部分修訂額定值的泄漏率不得大于25.0%,KF94額定值的泄漏率不得大于11.0%,KF99額定值的泄漏率不得大于5.0%。口罩的額定值(如:KF80、KF90、K99)應在產(chǎn)品名稱括號中注明。
衛(wèi)生口罩需并在成品上注明“按下列試驗方法進行試驗時,各單項測量值不應小于OOO%。”并在描述面部吸入阻力試驗測試方法時說明以下細節(jié):“將標準頭部模型放置在面部區(qū)域后進行試驗。使用六個衛(wèi)生口罩時,三個應與提交的一樣,另外三個應在溫度38±2.5℃和濕度85±5%RH下無人看管24±1小時,然后用作試樣。當空氣以每分鐘30升的連續(xù)流量噴過面部區(qū)域時,應測量水柱(mmH2O),并詳細說明試驗方法的其他細節(jié)。”
根據(jù)2014年9月4日的補充規(guī)定2014-153號,原本作為防塵口罩或防病口罩的產(chǎn)品,被允許作為公共衛(wèi)生口罩。
如果得到韓國食藥廳許可,口罩會在包裝上標上 “????(醫(yī)藥輔品)”。“???(手術(shù)用)”和“???(衛(wèi)生保健用)”都屬于醫(yī)藥輔品。
根據(jù)韓國海關(guān)官網(wǎng)2020年3月5日信息,口罩進口清關(guān)流程如下:
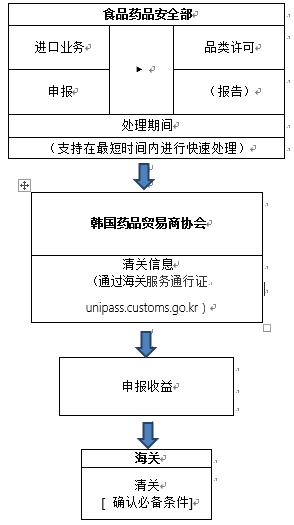
目前食品藥品安全部正在進行快速許可,許可審查部門的聯(lián)系方式如下:(聯(lián)系以獲取進口業(yè)務報關(guān)單和產(chǎn)品授權(quán)書,并迅速處理進口要求。 )
澳大利亞
1、必要資料:提單,箱單,發(fā)票。
2、須通過澳洲的TGA注冊,符合標準規(guī)范:AS/NZS 1716:2012,此規(guī)范是澳大利亞和新西蘭的呼吸保護裝置標準。
TGA 是Therapeutic Goods Administration的簡寫,全稱是治療商品管理局,它是澳大利亞的治療商品(包括藥物、醫(yī)療器械、基因科技和血液制品)的監(jiān)督機構(gòu)。澳大利亞對醫(yī)療器械分為I類,Is and Im, IIa, IIb, III類,產(chǎn)品的分類幾乎和歐盟分類一致,如果產(chǎn)品已經(jīng)獲得CE標志,則產(chǎn)品類別可以按照CE分類。
3、監(jiān)管及出口須知:
澳洲的醫(yī)用口罩按照I類管理,需要在TGA進行備案之后銷售。在澳洲的備案需要由澳洲當?shù)氐腟PONSOR來完成,其合規(guī)流程為:
① 指定SPONSOR;
只有通過澳大利亞的代理人才能夠提出申請,代理人這里有一個專有名詞叫“Sponsor”,簡單來說就是進口商。
② 完成技術(shù)文檔;
低風險的I類器械沒有強制性質(zhì)量體系和上市前評價的明確要求, 但要求制造商提供相關(guān)文件證明其安全有效性。
③ 提交TGA進行備案;
④ 獲得證書。
特別提醒:澳大利亞已與歐盟達成互認協(xié)議。這意味著,合格評定證書由TGA頒發(fā)的也被歐盟認可,TGA也認可歐盟CE認證。已獲CE認證的用戶,可提交CE證書及相關(guān)資料,獲得TGA證書。
提示:以上內(nèi)容是根據(jù)國內(nèi)外相關(guān)政府機構(gòu)、專業(yè)網(wǎng)站、新聞報道收集整理而成,僅供參考。具體內(nèi)容以相關(guān)管理部門、國外官方機構(gòu)要求為準。(來源:阿里巴巴外貿(mào)圈)
以上內(nèi)容屬作者個人觀點,不代表雨果網(wǎng)立場!如有侵權(quán),請聯(lián)系我們。



















